SMA type 1 is a severe inherited neuromuscular disease that causes rapid loss of motor neurons, leading to profound muscle weakness in infancy. It is caused by missing or nonworking copies of the SMN1 gene. Treatment options now include a one-time gene therapy and two SMN-boosting medicines, and outcomes are significantly better when therapy starts before symptoms appear.
What is SMA type 1?
Spinal muscular atrophy (SMA) is a group of genetic disorders. Type 1, also called Werdnig–Hoffmann disease, is the most common and most severe form that presents in infancy. It results from biallelic pathogenic variants in SMN1, which deprive motor neurons of the survival motor neuron (SMN) protein. Without adequate SMN, motor neurons in the spinal cord degenerate, causing progressive weakness and respiratory failure (GeneReviews).
SMA type 1 is a 5q-SMA caused by loss of SMN1 function. Severity is partly modified by the number of backup SMN2 gene copies, with fewer copies linked to more severe disease.
How does SMA type 1 affect babies?
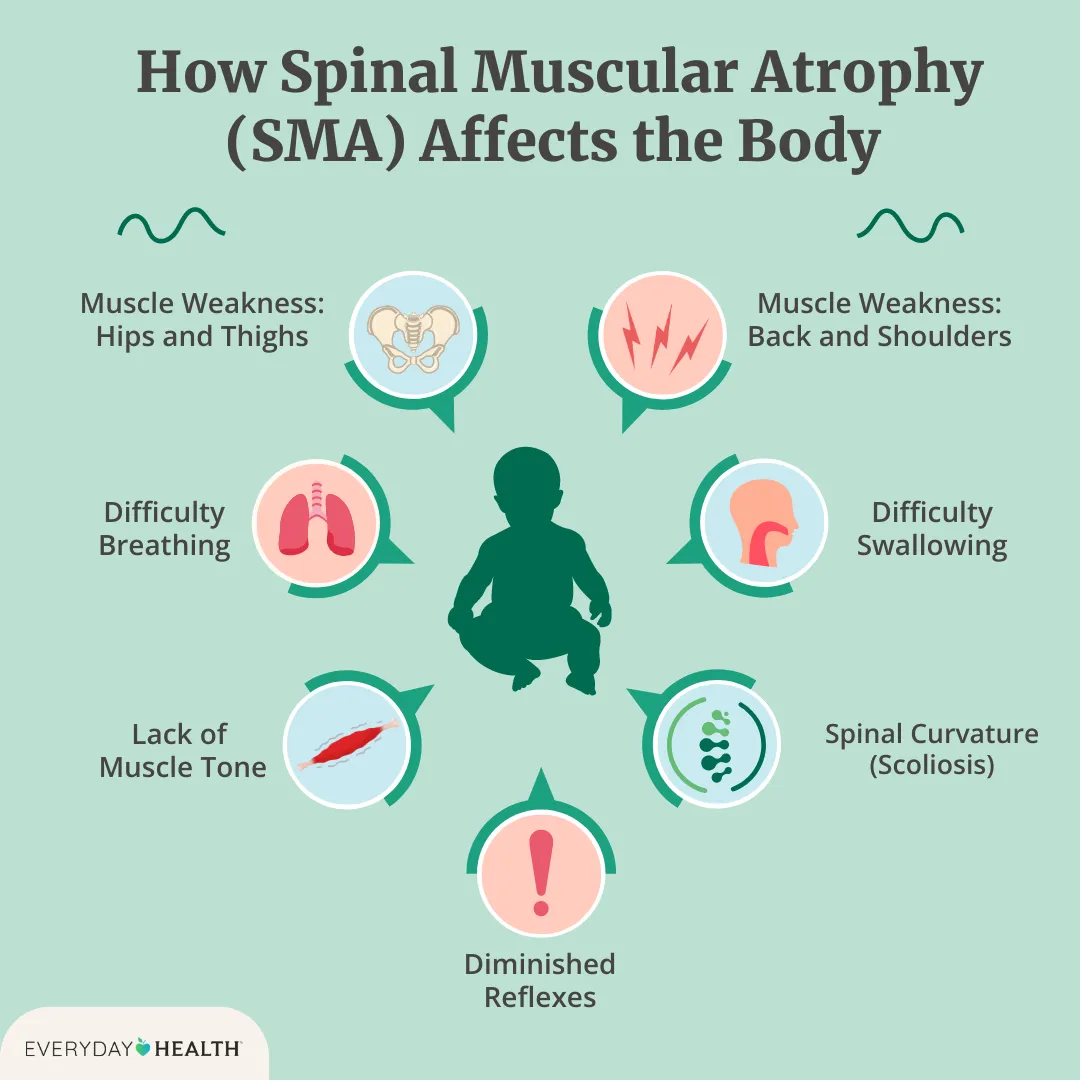
Symptoms typically appear before 6 months of age. Early signs include weak cry, poor head control, difficulty sucking or swallowing, diminished limb movement, hypotonia (floppiness), and paradoxical or labored breathing. Without treatment, most infants never sit independently, and many require permanent ventilation or die within the first two years (NINDS), (GeneReviews).
- Onset: birth to 6 months
- Motor milestones: typically cannot sit unsupported without treatment
- Breathing: progressive weakness of respiratory muscles
- Feeding: risk of aspiration and failure to thrive
Cognition is usually normal in SMA. The condition primarily affects voluntary muscles, not intellectual development (NINDS).
How is SMA type 1 diagnosed, and is there newborn screening?
Diagnosis is made with a blood test that looks for SMN1 deletions or mutations and determines SMN2 copy number. Electromyography or muscle biopsy is rarely needed today because genetic testing is definitive (GeneReviews).
Many countries now include SMA on their newborn screening panels because early treatment improves outcomes. In the United States, SMA is on the Recommended Uniform Screening Panel and is screened in nearly all jurisdictions (CDC).
In the United Kingdom, the routine NHS newborn blood spot test currently screens for nine conditions and does not include SMA (NHS). However, the NHS and Genomics England are evaluating genomic screening at birth in the Newborn Genomes Programme (Generation Study), which is researching whether actionable rare conditions such as SMA can be identified and treated earlier (Genomics England).
What treatments are available for SMA type 1 and how do they work?
Three disease-modifying therapies target the SMN pathway. Choice depends on age, clinical status, access, and clinician and family preference.
- Onasemnogene abeparvovec (Zolgensma): a one-time intravenous gene therapy using an adeno-associated virus vector to deliver a working SMN1 gene, enabling cells to produce SMN protein. It requires corticosteroid coverage and careful monitoring for liver injury, thrombocytopenia, and cardiac effects (EMA), (NHS).
- Nusinersen (Spinraza): an intrathecal antisense oligonucleotide that alters SMN2 splicing to increase full-length SMN protein. It is given by lumbar puncture with loading and maintenance doses. Clinical trials demonstrated improved survival and motor function in infants with SMA (NICE), (NHS).
- Risdiplam (Evrysdi): an oral small-molecule splicing modifier that increases SMN protein from SMN2. It is taken daily and has shown motor gains and survival benefits in infants and children with SMA (NHS).
Supportive care remains essential for all children with SMA. This includes respiratory support and airway clearance, nutritional management and swallowing safety, physiotherapy, orthopedic care, and immunizations including respiratory syncytial virus prophylaxis as appropriate (GeneReviews).
Why does early treatment change outcomes?
Because motor neuron loss begins before symptoms are obvious, acting early preserves function. Trials of presymptomatic infants treated shortly after birth show markedly better outcomes than in historically untreated cohorts.
- Gene therapy presymptomatic: Many infants achieved sitting and some achieved independent walking in the SPR1NT study of onasemnogene abeparvovec (Nat Med 2022).
- Nusinersen presymptomatic: The NURTURE study reported survival without permanent ventilation and attainment of motor milestones not seen in untreated SMA type 1 (Neurology 2021).
The best outcomes occur when therapy starts before or at the very first signs of weakness, which is why newborn screening and rapid confirmatory testing are so important.
What is the outlook today, and what are the limitations?
Outcomes for SMA type 1 have improved substantially with modern therapies, especially when started presymptomatically. Many treated children reach milestones such as sitting and, in some cases, standing or walking. When treatment begins after significant weakness is present, gains are often more modest and intensive supportive care remains necessary (GeneReviews), (NHS).
Therapies have risks and require monitoring. Gene therapy can cause serious liver inflammation, low platelets, and elevated cardiac enzymes. Nusinersen involves repeat lumbar punctures. Risdiplam may cause gastrointestinal and other side effects. Access, timing, and eligibility criteria vary by country and health system, and long-term data are still accumulating (EMA), (NICE).
Families should work with a multidisciplinary neuromuscular team to select and sequence therapies, coordinate supportive care, and plan for transitions as the child grows.